
YASARA よくある質問
YASARA TOPはこちら
目次
- Q1. YASARAにおいて重要な10の単語(The ten magic words )
- Q2. キーボード・マウスの基本的な使い方は?
- Q3. “Hide”、”Switch off”、”Remove”、”Delete”の違いは?
- Q4. 二面角の変更の方法は?
- Q5. ファイルのロード方法は?
- Q6. ファイルのセーブ方法
- Q7. ユーザーマニュアルはありますか?
- Q8. YASARA関連論文の情報や参考になる技術資料などはありますか?
- Q9. YASARAを起動することができません。何か対処法がありますか?
- Q10. Windows 7上でもYASARAを利用できますか?
- Q11. 金属イオンの結合部位を予測することができますか?
- Q12. 複数化合物のドッキングスクリーニングを実行するには?
- Q13. アンサンブルドッキングとスクリーニングを同時にできるマクロファイルはありますか?
- Q14. Bindenergy値は正に大きい方が結合が強いのですか?
- Q15. タンパク質のアミノ酸を置換や修飾することはできますか?
- Q16. YASARAの新バージョンや別OSのインストーラを入手するには?
- Q17. オブジェクトやSceneの前後(画面の手前や奥)を不可視化して、中心部分をスライスして表示するような機能はありますか?
- Q18. MDシミュレーションの実行時に、電界を設定することはできますか?
- Q19. 「Clean」や「OptHyd」コマンドを実行した際、意図しない箇所が自動的にプロトン化されてしまいます。
- Q20. 自作の力場パラメータを使って、力場の修正ができますか?
- Q21. プロテインモデリングの実行時に、機密配列が外部のサーバーに送信されますか。
- Q22. YASARAからAlphaFoldなどのAIベースのフォールディング手法を利用できますか。
- Q23. macOSでのYASARAのインストール方法やアプリケーションの起動方法を教えてください。
- Q24. マクロをYASARA GUIのコンソールに直接ペーストして実行することはできますか。
- Q25. MDトラジェクトリファイル~.simを開くには?
- Q26. 相互作用の色の割り当ては?
- Q27. 結合次数を確認する方法は?
- Q28. MD解析レポートのグラフやプロットの数値データはどこから取得できますか?
Q1. YASARAにおいて重要な10の単語(The ten magic words )
【1】Atom
YASARAにおける原子の表現。
各Atomには元素名の他に固有の名前(Name,最大4文字)と番号(Number)が振られている。
また、各Atomが含まれているResidue, Molecule, Objectの情報も記載されている。
【2】 Residue
YASARAにおける残基(原子の連続的な集合体)の表現。各ResidueにAtomが格納されている。
各Residueには固有の名前(Name,最大3文字)と番号及びコード(Number, Insertion code)が振られている。
【3】 Molecule
YASARAにおける分子(残基の連続的な集合体)の表現。各MoleculeにResidueが格納されている。
各Moleculeには固有の名前(Name,最大1文字)が振られている。
【4】 Object
Molecule 及び追加アイテムの集合体を表す(連続している必要なし)。各ObjectにMoleculeが格納されている。
マウスやキーボードを用いて移動させることができる。
追加アイテムとしては矢印やラベルなどの情報が含まれる。
アクティブ状態と非アクティブ状態を切り替えることができ、非アクティブ状態のObjectはSoupから除かれる。
(切り替えは「Remove」コマンドと「Add」コマンドにて行う)
【5】Soup
アクティブなObjectに含まれるすべてのAtomを意味する。
矢印やラベルなどの情報は含まれない。
【6】Scene
アクティブであるか非アクティブであるかにかかわらず、すべてのObjectをひとまとめにしたものを意味する。
Sceneとして保存した場合、矢印やラベルなどを含む全Objectについて、位置や向きも維持したまま保存することができる。
【7】All
コマンドを実行するにあたって指定可能なすべての対象を意味する。
この時”All”が「全てのObject(Scene)」を指すか、「アクティブなObject中の全てのAtom(Soup)」を指すかは、各コマンドによって異なる。
【8】Bond
pH依存性に従って決定される2原子間の共有結合のこと。
化学的な「結合」には様々な種類が存在するが、YASARAでは典型的な共有結合のみに”Bond”という語を用いる。
イオン結合や配位結合などは結合(Bond)として処理されない。分子動力学計算において力場(force field)はこれらの結合を再現するために静電相互作用を考慮するので、”Bond”として明示的に結合を加えることは力場パラメータを割り当てる際に問題を起こす原因となる。
PDBファイルにて金属イオンとの結合が指定されている(‘CONECT’から始まる行で記されている)場合、YASARAはそれを”Arrow bond”(矢印)に変換する。灰色の円柱として可視化できるようになるが、Soupには含まれないため、金属結合の構造を保持するためにシミュレーション開始時に距離と角度の拘束を与えるという一つ例外を除き、ほとんどの計算では無視される。
共有結合には単結合や二重結合など複数の次数(Order)が存在するが、YASARAでは共鳴構造も含めて非整数の結合次数(fractional bond orders)もサポートされている。これにより対称性の保持や潜在的な化学的性質をより良く表すことができる。<F2>キーを押してSceneスタイルを”ball & stick”に変更し、View > Color > Bonds からカラースケールで’Order’を選択すると、下表のように色分けがなされ、視覚化することができる。
| Name | 結合次数(Order) | 色(Color) |
|---|---|---|
| 単結合(Single bond) | 1 | グレー(gray) |
| 共鳴(Resonance bond) | 1.25 | 青(blue) |
| 共鳴(Resonance bond) | 1.33 | マジェンタ(magenta) |
| 共鳴(Resonance bond) | 1.5 | 赤(red) |
| 共鳴(Resonance bond) | 1.66 | 明るいオレンジ(bright orange) |
| 二重結合(Double bond) | 2 | 黄(yellow) |
| 共鳴(Resonance bond) | 2.5 | ライムグリーン(lime green) |
| 三重結合(Triple bond) | 3 | 緑(green) |
| 四重結合(Quadruple bond) | 4 | シアン(cyan) |
また、結合次数はpH依存性に基づき割り当てられる。YASARAは、次の二つの方法でpHの影響を考慮している。
- 1.一般的なpHモデル
-
SMILES文字列のライブラリに基づき、現在設定されているpH(Default pH)に従って、結合次数の割り当て、水素原子の追加を行う。
Default pHはOptions > Default pHで変更可能。変更の際に’Adjust bond orders and hydrogens’ボックスをチェックすると、変更後のpHに適した状態になるようにすべてのBondの結合次数および水素の再割り当てが自動で行われる。
SMILES文字列ライブラリには、「カルボキシル基はph3以下で中性、それ以上では負に帯電する」等の記載がされている。
詳細は、ファイル”yasara.def”の’GROUP_DATA’セクションを参照。 - 2.アミノ酸専用pHモデル
-
分子動力学シミュレーション等を行う場合、上記の一般的pHモデルでは不十分である。
特に活性部位の残基は環境に応じて大きなpKaシフトを示すことが多く、「カルボキシル基はph3以下で中性である」という一般的モデルに代わって、「残基Glu 42のpKaが5.1に上昇し、プロトンはOE1の酸素に存在しやすい」というような特別なモデルが選択される。
これらのpKaの予測は、分子動力学シミュレーションの準備として用いられる「細胞中和実験(cell neutralization experiment)」によって行われる。なお、pKaResコマンドを用いて実験的に測定したpKa値に上書きすることも可能である。
まず初めに Edit> Clean> All 等の操作を行って一般的pHモデルを適用し、その後Option > Choose experiment > Cell neutralization 等の操作で細胞中和実験を行うのがよい。細胞中和実験完了後は、特殊な調整がなされていることが考慮されないため、再び一般的pHモデルを適応させないように注意すること。
次に、一般的pHモデルの概念をいくつかの例で示す。
Figure: pHの値に基づく結合次数と水素付加パターン
(protonation patterns)
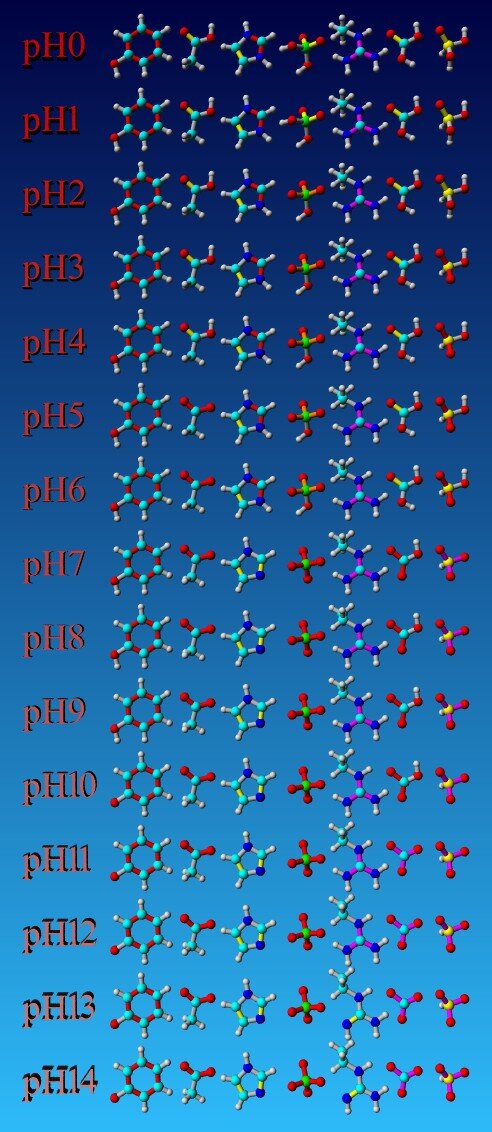
上図はpH0~14の環境における7種の低分子の様子を示したものである。左から順に解説する。
- 1.フェノール
- 芳香環のC-C結合は全てほぼ同等であり、結合次数は1.5(赤)である。ph20以上でヒドロキシル基はプロトンを失う。
- 2.酢酸
- pHが0から4の場合、カルボキシル基は中性であり、一方の酸素は二重結合(黄)を示し、他方には水素が付加されている。pH5以上ではプロトンがなくなり、両方の酸素は等価となって次数1.5(赤)の結合を形成する。
- 3.イミダゾール環
- pH6が0から6の場合、環がプロトン化され、2つの窒素がそれぞれ1つの水素を持ち、中間の炭素と次数1.5の等価な結合を作る。したがって、窒素の原子価は3.5となり、個々の窒素の形式電荷は+0.5、合計で+1となる。pH7 以上では対称性が損なわれ、分子は中性となり、水素が脱離した窒素が二重結合を形成する。
- 4.硫酸
- ph2未満では、分子は中性であり、2つの酸素は二重結合を形成し、他の2つは水素を保持する。 ph2~6では水素1個が脱離し、3つの酸素は共鳴効果により等価となって、それぞれ次数1.66(オレンジ)の結合を形成する。硫黄原子の原子価は6(1 + 3×1.66)となり、3つの酸素それぞれに生じる形式電荷は-0.33、合計-1になる。 pH7以上では、両方の水素が分離し、4つの酸素は全て1.5の等価結合を形成する。結果として生じる4つの形式電荷は-0.5、合計して-2になる。
- 5.グアニジウム基
- アルギニンの側鎖の一部を形成する官能基であり、共鳴効果のために強い塩基となっている。 ph22以下のとき、基はプロトン化され、各窒素は中心炭素に次数1.33(マゼンタ)の結合を形成する。したがって、炭素原子価は通常のまま(4)であり、各窒素は3.33の原子価を有し、個々の形式電荷は+0.33、合計で+ 1になる。 pHが13になると、対称性が損なわれ、1つの窒素がプロトンを失い、二重結合を形成する。
- 6.炭酸
- pH7未満では、分子は中性であり、一方の酸素は二重結合を形成し、他の2つは水素を持つ。 pH 7~10では、水素1個がなくなり、2個の酸素は次数1.5の等価結合(赤色)を形成する。 ph21以上の場合、両方の水素が解離し、3つの次数1.33(マゼンタ)の等価結合となる。酸素3つの形式電荷は-0.66、合計で-2となる。
- 7.ホスホン酸
- 炭酸と同様。リンは価数が5であるので、追加の水素原子を有する。
【9】Local coordinate system
個々のObjectに適応される座標系のこと。
一般的な分子視覚化プログラムは単一座標系で動作するものが多く、複数のPDBファイルをロードしてそれを個別に移動したり回転させたりすると、不具合が生じる場合がある。
YASARAでは、すべてのObjectが独自のLocal coordinate system(ローカル座標系)を持つ。ローカル座標系はPDBファイルで指定されるデカルト座標(直交座標)に対応している。
ファイル読み込みの際に’Center’オプションを無効にして(File > Load からロードする場合は右上の’Center object’ボックスのチェックを外して)ロードを行うと、個々のAtomのローカル座標はPDBファイルのデカルト座標と完全に一致する(左手座標系の場合はX軸のパラメータの正負が反転)。
Objectを’Center’に設定すると座標が移動し、左手座標系を使用するとX座標の符号が反転するが、Objectの移動または回転によって原子座標が変化することはない。
ただし、場合によっては複数のObjectのローカル座標系が一致することが望ましい場合もある。ローカル座標系を一致させる方法としては以下のような手法が挙げられる。
- 1.’Center’オプションを無効にして、Objectを互いに重なり合わせてロードする
- (重ね合わせがなされるまでObjectは移動させないこと)
- 2.Transferコマンドを用いて、あるObjectを他のObjectの座標系に移動する。
- ※互いに関係するObjectの座標系を一致させる場合には上記二つの方法が有効
- 3.Superposeコマンドを用いて、Objectを重ね合わせる。
- 4.Joinコマンドを用いて、Objectを統合する。
- 5.Transformコマンドを用いて、Objectの座標系を変換する。
- 6.シミュレーションを開始して、Objectをシミュレーションセルの座標系に転送する。
同様に、相対位置を反映する原子座標を持つ2つのObjectを独立して保存する場合は、ローカル座標系が一致すること、すなわち2つのObjectが同じグローバル座標を持つことを確認する必要がある。
Edit > Transfer > Objectを選択し、1つ目のObjectを指定、次に2つ目のObjectを指定、最後に上から3番目のオプション’Fix atoms on screen during the transfer…’を選択すると、両方のObjectが同じグローバル座標を共有する。
※PDBファイルを保存する場合には、’Transform’ボックスのチェックを外す必要がある。
【10】Global coordinate system
Scene全体に適応される座標系のこと。
すべてのObjectは、YASARAのGlobal coordinate system(グローバル座標系)によって配置・方向付けがなされる。
これらのグローバル座標はObjectを移動または回転させると変化する。
単一のObjectを指定しない限りは、すべてのObjectは一つの座標を共通の中心として回転する。
下図はローカル原子座標からグローバル画面座標を生成する方法を図解したものである。
- Objectはまず原点(Screen Center,0/0/0)に配置される。
- 次にX,Y,Z軸それぞれについてAlpha,Beta,Gammaの角度で回転される。(回転の角度はOriObjコマンドか、マウスでの回転で変更が可能)
- 最後にグローバル位置(Global PosObj,デフォルト0/0/50)に移動される。(グローバル位置はPosObjコマンドか、マウスでの移動で変更が可能)
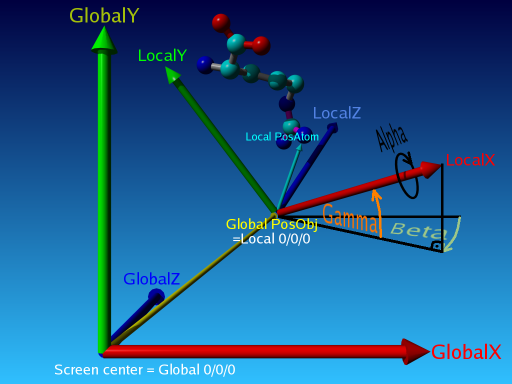
座標システムは左手座標系(left-handed coordinate system,デフォルト)と右手座標系(right-handed coordinate system)の両方を選択可能。
座標システムの切り替えは Option > Coordinate system より変更できる。(切替えの際にはClear処理(Sceneの削除)がされるので注意)

原子の周囲に球オブジェクト(Sphere)を発生させたい場合など、ローカル座標系だけが分かっている位置にグローバル座標系でObjectを配置したい場合には、以下のような方法を用いて移動先のグローバル座標を取得してObjectを配置する必要がある。
以下では例として、PDB:1crnのAtom 1上に球オブジェクトを発生させる手法(左手座標系の場合)を記す。
1.PosAtomコマンドに’CoordSys = Global’を指定してグローバル座標を取得し、その座標にObjectを移動する。
入力例(以下コンソール内):
LoadPDB 1crn.pdb PosAtom 1,CoordSys=global #出力 Atom 1 N THR 1 A global position: X=-7.778, Y=4.312, Z=46.658 A ShowSphere radius=3,color=blue,alpha=50 PosObj Sphere,X=-7.778, Y=4.312, Z=46.658
2.’Center’オプションを無効にして(メニューから選択する場合には’Center object’ボックスのチェックを外して)PDBファイルをロードすると、ローカル座標がPDBファイルのデカルト座標と一致する(左手座標系の場合はX軸のパラメータの正負が反転)。次いでObjectのグローバル位置を0/0/0に移動させると、グローバル座標をPDBファイルのデカルト座標およびローカル座標と一致させることができる。最後にPDBファイルのデカルト座標に合わせてObjectを移動する。
入力例(以下コンソール内):
#まずPDBファイルを確認してAtom1の座標を取得する。 #(PDBファイル内:ATOM 1 N THR 1 17.047(←X座標) 14.099(←Y座標) 3.625(←Z座標) 1.00 13.79 1CRN 70) #座標を入力する際、左手座標系ではX軸の符号が反転することに注意。 LoadPDB 1crn.pdb ,Center=No PosObj 1crn,X=0,Y=0,Z=0 ShowSphere radius=3,color=blue,alpha=50 PosObj Sphere,X=-17.047,Y=14.099,Z=3.625
(2020/10/1)
Q2. キーボード・マウスの基本的な使い方は?
※以下の内容は主にWindows版を対象としています。
Linux, Mac, Android版についてはユーザーマニュアルをご確認ください。
オブジェクトの移動
<Key>
| Shift | 移動させるObjectの変更(1つ進む [ 例:Obj all → Obj 1 → Obj 2 → Obj 3… ]) |
|---|---|
| Ctrl + Shift | 移動させるObjectの変更(1つ戻る) |
| カーソルキー | X/Y軸方向へのObjectの移動 |
※以下のキーは低分子buildモードでは使えません。
| 1~9 (テンキー不可) | Object及びScene移動速度の変更(1:遅~9:速) |
|---|---|
| Q,W,E,R,T,Y | Object及びSceneの移動(Q・W=X軸方向,E・R=Y軸方向,T・Y=Z軸方向) |
| Ctrl+1~9 (テンキー不可) | Object及びScene回転速度の変更(1:遅~9:速) |
| A,S,D,F,G,H | Object及びSceneの回転(A・S=X軸中心,D・F=Y軸中心,G・H=Z軸中心) |
| Z,X,C,V,B,N | Object及びSceneの自動回転(Z・X=X軸中心,C・V=Y軸中心,B・N=Z軸中心) |
<マウス>
| 左ドラッグ + マウス移動 | Object及びSceneのX/Y軸方向での回転 |
|---|---|
| Ctrl + 左ドラッグ + マウス移動 | 選択中の原子を中心としたObject及びSceneのX/Y軸方向での回転 |
| 左右ドラッグ(同時) + マウス移動 | Object及びSceneのX/Y軸方向への移動(※ホイールボタンをドラッグしても同様) |
| 右ドラッグ + マウス移動 | Object及びSceneのZ軸方向への移動 |
| [ラベル上で]左クリック | ラベルの選択 |
| Ctrl + 右ドラッグ + マウス移動 | マークされたラベルのサイズ変更 |
| [ラベル上で]ホイールボタンクリック | ラベルの向きをデフォルトにリセット |
Sceneスタイルの変更
| F1 or Alt+1 | Sceneスタイルを”ball”に変更 |
|---|---|
| F2 or Alt+2 | Sceneスタイルを”ball & stick”に変更 |
| F3 or Alt+3 | Sceneスタイルを”stick”に変更 |
| F4 or Alt+4 | Sceneスタイルを”Calpha trace”に変更 |
| F5 or Alt+5 | Sceneスタイルを”tube”に変更 |
| F6 or Alt+6 | Sceneスタイルを”ribbon”に変更 |
| F7 or Alt+7 | Sceneスタイルを”cartoon”に変更 |
| F8 or Alt+8 | 側鎖の表示方法切り替え [ 例:hide → stick → ball&stick ] |
| HUD上の”Visibility”列を左クリック | クリックしたObject等の表示・非表示の切り替え |
| HUD上の”Visibility”列を右クリック | クリックしたObject等のみを表示し、その他はすべて非表示に変更 |
編集
| Delete | マークしたAtomやラベルの削除 |
|---|---|
| Alt+左クリック(個別のAtom) | 最後のコマンドの繰り返し(デフォルトがKDEの場合は代わりにTabを使用) |
| Alt+左クリック(sequence selector上) | 最後のコマンドの繰り返し(デフォルトがKDEの場合は代わりにTabを使用) |
| Ctrl+Z or Ctrl+U | 操作を一つ戻す(Undo) |
| Ctrl+R | 操作を一つ進める(Redo) |
※sequence selector:YASARA操作画面の下部にカーソルを合わせると表示されるResidue一覧のこと。
Soupの分析
| Insert or I | HUD(head up display)の表示・非表示切り替え |
|---|---|
| Ctrl+Insert or Ctrl+I | 右HUDの表示情報切り替え |
| 左クリック(個別のAtom) | Atomをマークする(HUDにプロパティが表示される) |
| 左クリック(sequence selector上) | クリックしたResidue中のAtomをマークする |
| PageDown or ホイールボタン下回転 | 現在マークしているAtomの、一つ次の番号のAtomをマークする |
| PageUp or ホイールボタン上回転 | 現在マークしているAtomの、一つ前の番号のAtomをマークする |
| Ctrl+左クリック(複数のAtom) | マークされた原子の距離(distance)・角度(angle)・二面角(dihedral)の情報を表示 |
| Ctrl+左クリック(sequence selector上) | クリックしたResidueへズームする |
| Ctrl+右クリック(個別のAtom) | Atom向けコンテクストメニューの表示 |
| Ctrl+右クリック(sequence selector上) | Residue向けコンテクストメニューの表示 |
| Home | 一つ前の charge 及び bond の概要を表示する(simulation HUD) |
| End | 一つ後の charge 及び bond の概要を表示する(simulation HUD) |
| [sequence selector上]任意の英字キー | 入力された文字(1文字)に対応するアミノ酸をResidue中から探索する (ポインタがsequence selector上にある時のみ有効) |
| [sequence selector上]Ctrl+V | クリップボード上の文字に対応するアミノ酸をResidue中から探索する (ポインタがsequence selector上にある時のみ有効) |
※sequence selector:YASARA操作画面の下部にカーソルを合わせると表示されるResidue一覧のこと。
低分子Buildingモード
![]()
YASARAトップメニューにあるベンゼン環の形をしたボタンをクリックすると低分子Buildingモードに切り替わり、以下のキー操作が有効になります。(水素の付加や結合距離などは自動的に調整されます。)
| H | マークされているAtomを「水素(hydrogen)」に変更 |
|---|---|
| C | マークされているAtomを「炭素(carbon)」に変更 |
| N | マークされているAtomを「窒素(nitrogen)」に変更 |
| O | マークされているAtomを「酸素(oxygen)」に変更 |
| F | マークされているAtomを「フッ素(flourine)」に変更 |
| P | マークされているAtomを「リン(phosphorus)」に変更 |
| S | マークされているAtomを「硫黄(sulfur)」に変更 |
| I | マークされているAtomを「ヨウ素(iodine)」に変更 |
・2つのAtomをクリックで指定(マーク)している場合、以下の操作でBondの変更やブリッジ構造の作成ができます。
(水素の付加や結合距離などは自動的に調整されます。)
| 1 | マークされたAtom間のBondを”single bond”に変更 |
|---|---|
| 2 | マークされたAtom間のBondを”double bond”に変更 |
| 3 | マークされたAtom間のBondを”triple bond”に変更 |
| 4 | マークされたAtom間のBondを”quadruple bond”に変更 |
| 5 | マークされたAtom間のBondを”resonance bond with order 1.5″に変更 |
| C | マークされたAtom間に炭素ブリッジを作成 |
| N | マークされたAtom間に窒素ブリッジを作成 |
| O | マークされたAtom間に酸素ブリッジを作成 |
| P | マークされたAtom間にリンブリッジを作成 |
| S | マークされたAtom間に硫黄ブリッジを作成 |
シミュレーション制御
| F9 or Alt+9 | 全エネルギーの計算、及びHUD(simulation parameters)への結果表示 |
|---|---|
| F10 or 0 | シミュレーション温度管理の変更(一つ進む) |
| Ctrl+F10 or Ctrl+0 | シミュレーション温度管理の変更(一つ戻る) |
| F11 or 0の一つ右側のキー | シミュレーションの一時停止 |
| Ctrl+F11 | 一時停止中のシミュレーションを1ステップだけ進める |
| F12 or 0の二つ右側のキー | シミュレーションの実行・停止の切り替え |
| 1~9 (テンキー不可) | 引き寄せる強さの変更(1:弱~9:強) |
| Ctrl+右ドラッグ | マークしている原子をマウスポインタの方向に引き寄せる(シミュレーション中) |
| Ctrl+左右ドラッグ | マークしている原子を含むObjectをマウスポインタの方向に引き寄せる(シミュレーション中) |
| Ctrl+ ホイールボタンドラッグ | マークしている原子を含むObjectをマウスポインタの方向に引き寄せる(シミュレーション中) |
コンソールの使用
| スペースキー | コンソールを展開する |
|---|---|
| スペースキー(二回連続) | コンソール画面をフルサイズにする |
| リターン(エンター)キー | (何も入力していない状態の場合のみ)コンソールを閉じる |
| マウス左ボタン | テキストの選択、およびクリップボードへのコピー |
| Ctrl+A | 全テキストの選択、およびクリップボードへのコピー |
| ホイールボタンクリック | クリップボードのテキストを貼り付ける |
| Ctrl+V | クリップボードのテキストを貼り付ける |
| PageUp | コンソールを1ページ下げる |
| PageDown | コンソールを1ページ上げる |
| ホイールボタン上回転 | コンソールを半ページ下げる |
| ホイールボタン下回転 | コンソールを半ページ上げる |
| Ctrl+上カーソルキー | コンソールを一行だけ下げる |
| Ctrl+下カーソルキー | コンソールを一行だけ上げる |
| 上カーソルキー | コマンド履歴から一つ前のものを表示する |
| 下カーソルキー | コマンド履歴から一つ後のものを表示する |
| Home | カーソルをラインの最初に移動 |
| End | カーソルをラインの最後に移動 |
| Ins | HUDの表示・非表示切り替え |
| Ctrl+Ins | HUDの表示項目切り替え |
その他
| Pause or P | YASARAの一時停止 |
|---|---|
| Ctrl+Pause or Ctrl+P | 一時停止中のYASARAを1ステップだけ進める |
| Ctrl+M | 最後のマクロまたはムービーを再実行 |
トップメニュー上のボタンについて
![]()
| 低分子Buildingモードへの切り替え | |
| 長方形状にAtomを選択 | |
| 輪状にAtomを選択 | |
| 操作を一つ戻す | |
| 操作を一つ進める | |
| Soupをすべて削除し、新しいSceneを始める | |
| Sceneファイルを開く | |
| Sceneファイルを上書き保存する(Sceneの読み込みや保存を行った後のみ有効) | |
| Sceneを画面いっぱいに収まるように移動させる | |
| ラベルが含まれているマクロやムービーを、一つ前のラベルに戻す | |
| ラベルが含まれているマクロやムービーを、一つ次のラベルに進める | |
| Z軸中心でSceneを回転させる |
(2020/10/1)
Q3. “Hide”、”Switch off”、”Remove”、”Delete”の違いは?
YASARAにおいて表示されている化合物を「消す」作業を行う場合、いくつかのコマンドの中から自分の目的に沿ったコマンドを用いる必要があります。
それぞれのコマンドには以下のような違いが存在します。
Hide
選択したAtomがYASARAの画面上に表示されなくなります。
“Hide”の効果を受けるのは個々のAtomですが、ObjectやMoleculeの単位でいくつかのAtomを選択することもできます。
再び表示されるまでは画面に映りませんが、Objectの一部として残っており、スタイルの変更も可能です。
再表示は”Show”で行います。
Switch off
選択したObjectがYASARAの画面上に表示されなくなります。
Atomを非表示に切り替える”Hide”に近いですが、Objectには矢印やラベルなどの情報も含まれるため、これらも非表示にすることができます。
Objectの表示はAtomの表示よりも上位のため、”Switch off”されたObject中のAtomに”Hide”,”Show”を行っても表示は切り替わりません。
再び表示されるまでは画面に映りませんが、Soupの一部として残っているため、シミュレーションなどは有効です。
再表示は”Switch on”で行います。
Remove
選択したObjectを非アクティブ状態にします。
非アクティブ状態にされたObjectはSoupから除外され、画面上にも表示されなくなります。
Sceneの中にデータは残りますが、アクティブ状態に切り替わるまではシミュレーション等の操作の対象にすることができなくなります。
アクティブ状態への切り替えは”Add”(Edit > Add > Object)で行います。
Delete
選択したObjectおよびAtomをYASARAから完全に削除します。
Sceneのどこにもデータが残らないため、表示もされず、操作の対象にもできず、保存することもできなくなります。
もしも重要な部分を”Delete”してしまった場合は、”Undo”で一つ操作を戻すことによって再度表示させることができる場合もありますが、複雑な操作の後などには”Undo”で戻れなくなる場合もあるため、注意が必要です。
(2020/10/1)
Q4. 二面角の変更の方法は?
分子の二面角の調整・変更は、「Dihedral」コマンドから実行することができます。
アラニンを例にとって、下図のAtom①②③平面とAtom②③④平面との二面角を変更する(=①②間の結合を中心に回転する)手法を見ていきましょう。
(なお今回の例ではBuildコマンドを用いてアラニンを生成しています。)
まずはCtrl+クリックでAtomをマークしていきます。
①、②、③、④の順でAtomをマークすると、Atom①②③平面が固定され、Atom②③④平面が回転することになります。
4つの原子をマークしたら、次にAtom①の上で右クリックしてメニューを表示させます。
メニューから、Geometry > Dihedral を選択してください。
すると、下図のようなウィンドウが現れます。
バーを左右に移動させることで二面角を回転させることができます。
また、画面上部トップメニューから「Dihedral」コマンドを実行させることでも二面角の変更は可能です。
Edit > Geometry > Dihedral を選択すると下図のようなウィンドウが現れます。
ここで4つのAtomを選択していきますが、今回はAtomの名前を指定する必要があります。
Atom名は画面右側HUDから確認することが可能です。また、分子をマークすると画面左側HUDにも表示されます。
(なお、Atomの選択の際には名前や元素などから複数のAtomを選択することもできますが、ここでは説明を省略します。)
4つのAtomを指定した後は二面角の数値を入力してOKをクリックすることで二面角を変更できます。
(Consider only bound atomsをチェックすると化学的結合のある原子同士のみを対象にしますので、複数の二面角変更を一度に行う際に便利です。)
(2020/10/1)
Q5. ファイルのロード方法は?
YASARA独自のファイル形式(“.yob”,”.sce”)
YASARA Object File (.yob)
YASARA Object (YOB) ファイルとは、YASARAで迅速に処理できるように記述されたバイナリファイル形式です。
一つのObjectだけを含む形式で、同等のPDB形式ファイルよりもサイズが50%ほど小さくなっています。
また、PDB形式では保存できないラベルや矢印などの情報も同時に保存することができます。
YOBファイルのロードは、メニューから File > Load > YASARA Object を選択します。
YASARA Scene File (.sce)
YASARA Scene (SCE) ファイルとは、YASARAのScene全体の情報を記載することのできるバイナリファイル形式です。
YOBファイル同様、同等のPDB形式ファイルよりもサイズが50%ほど小さくなっています。
一方でYOB形式とは異なり、複数のObjectを同じファイルに保存することが可能です。
複数の化合物の同一ファイルへの保存だけでなく、化合物とシミュレーションセルを同一のファイルに保存することもできるため、シミュレーション用マクロの実行などでも重要となるファイル形式です。
SCEファイルのロードは、メニューから File > Load > YASARA Scene を選択します。
PDBファイルのロード
GUIからYASARAにPDB形式ファイル(.pdb)を読み込ませる方法としては以下のような方法が存在します。
Load PDB file — 直接PDBファイルをロード
メニューから、File > Load > PDB file を選択します。
コンピューター上のPDBファイルを直接指定します。
例)YASARAホームディレクトリ上の”pdb”ディレクトリ内に存在する”1crn.pdb”を開く場合
Load PDB file from local PDB — 圧縮ファイルからのロード
分子量の大きい構造の場合、PDB形式ではデータが大きいため、コンピューターの容量を圧迫してしまいます。
RCSBのHPなどでは、通常のpdbファイルと並んでgzip形式で圧縮されたもの(.pdb.gz)が保存されています。
これらをダウンロードした後YASARAで利用する際、YASARAではgzipファイルを展開させることなく、PDBファイルを読み込むことが可能です。
(なお、gzip形式の他、tar形式にも対応しています。)
メニューから、 File > Load > PDB file from local PDB を選択します。
ダイアログが表示されたら、読み取るPDBのIDと、gzipで圧縮されたPDBファイルへのパスを入力します。
パスを入力する際、必ずファイル名のPDB IDの部分が”????”という形になるよう、”?”(クエスチョンマーク)を4回繰り返して入力してください。
※”????”がパスに含まれていない場合、エラー表示がされてPDBの読み込みは行われません。ファイル名にPDB IDが含まれていない場合は、PDB IDの項目を空欄とし、パスの適当な部分に”????”を入力してください。
例)YASARAホームディレクトリ上の”pdb”ディレクトリ内に存在する”1abs.pdb.gz”を開く場合
Load PDB file from Internet — WEBデータベースからのロード
YASARAではWEB上のデータベースから直接PDBを指定してダウンロードすることも可能です。
ダウンロード先として選択可能なのは以下の2サイトです。
・RCSB
・PDB_REDO
メニューから、 File > Load > PDB file from Internet を選択します。
ダイアログが表示されたら、PDB IDとダウンロード先のサイトを選択してOKをクリックします。
なおデフォルトの状態であれば、一度WEBデータベースからダウンロードされたPDBファイルは、自動的にYASARAホームディレクトリ上の”pdb”ディレクトリ内にgzipで圧縮された状態で保存されます。
※上の”PDB file from local PDB”のパスを変更すると、gzipファイルの保存先が変更されます。
例)RCSB.orgから1crnをダウンロードする場合
その他の形式のファイルのロード
PDBx/mmCIF形式
従来のPDB形式では、ファイル自体の複雑さや記載できる分子の大きさに限界のあることが問題となってきており、近年新しいフォーマットとしてPDBx/mmCIF形式が普及され始めています。
(PDBx/mmCIF形式の詳細については、PDBjのサイト(https://pdbj.org/info/new-format)などをご参照ください。)
YASARAもPDBx/mmCIF形式に対応しており、ファイル変換の作業を行うことなくファイルを読み込むことが可能です。
メニューから、File > Load > PDBx/mmCIF fileを選択します。
ダイアログが表示されたら、コンピューター上のファイルを直接選択し、OKをクリックします。
その他の形式
YASARAは基本的にYASARA Object, YASARA Scene,
PDB(PDBx/mmCIF)以外のファイル形式には対応していませんが、Openbabelを利用して他の形式のファイルを変換することで、YASARAでも利用することができるようになります。
メニューから、File > Load > Other file format を選択します。
ダイアログが表示されたら、ファイルの形式とコンピューター上のファイルを両方選択し、OKをクリックします。
(2020/10/1)
Q6. ファイルのセーブ方法
分子構造ファイルの保存はそれぞれのファイル形式ごとに別のコマンドを用いる必要があります。
個々のファイル形式毎の保存方法は以下のとおりです。
YASARA Scene
SCEファイルのセーブは、メニューから File > Save as > YASARA Scene を選択します。
現在Scene上に存在するすべてのObjectが一つのSCEファイルにまとめて保存されます。
ダイアログが表示されたら、保存先とファイル名を選択して保存します。
YASARA Object
YOBファイルのセーブは、メニューから File > Save as > YASARA Object を選択します。
YOBファイルでは一つのObjectしか保存することができませんので、Objectが複数ある場合には複数のファイルを作成するか、一つのObjectにまとめるかする必要があります。
ダイアログが表示されたら、保存するObjectを指定し、保存先とファイル名を選択して保存します。
PDB、PDBx/mmCIF
PDBファイルのセーブは、メニューから File > Save as > PDB file を選択します。
PDBx/mmCIFファイルのセーブは、メニューから File > Save as > PDBx/mmCIF file を選択します。
単一のObjectが基本単位にはなりますが、複数のObjectを保存することも可能です。
ダイアログボックス上でCtrlキーやShiftキーを用いて複数のObjectを選択したり、Object名や分子の特徴によって選択したりすることができます。
ダイアログが表示されたら、保存するObjectを指定した後、保存先とファイル名を選択して保存します。
その他の形式
その他の形式でのセーブは、メニューから File > Save as > Other file format を選択します。
Openbabelを用いることで様々な形式のファイルを作成することが可能です。
ただし、単一のObjectしか保存することができません。
ダイアログが表示されたら、保存するObjectとファイルの形式を選択し、保存先とファイル名を選択して保存します。
(2020/10/1)
Q7. ユーザーマニュアルはありますか?
ソフトのユーザーマニュアルは、下記の方法によりご参照ください。
YASARA操作画面上部のメニューより、Help > Show user manual を選択します。
Webブラウザが起動され、ユーザーマニュアルを確認することができます。
また、このユーザーマニュアルの「Essentials > Getting started」内、箇条書きで「A single PDF file of this manual for your eBook reader can be found here.」と記載されている部分のリンクをクリックすると、PDF版のチュートリアルを開くこともできます。
なお、このPDFはYASRAインストールディレクトリ内に保存されています (yasara/doc/YASARAStructureManual.pdf)。
その他の参考資料や技術情報については、次の「Q8. YASARA関連論文の情報や参考になる技術資料などはありますか?」もあわせてご参照ください。
(2026/1/22)
Q8. YASARA関連論文の情報や参考になる技術資料などはありますか?
YASARA関連論文については、下記のリンクをご参照ください。
・YASARA開発者らによる文献リストはこちら
・YASARA引用文献リストはこちら
その他技術情報については、下記のチュートリアル資料や弊社ブログをご参照ください。
・各種チュートリアル紹介ページはこちら
(2024/4/9)
Q9. YASARAを起動することができません。何か対処法がありますか?
まず考えられるのは、グラフィックドライバの問題です。最新版ドライバへ更新いただいた後、YASARAの起動をお試しください。それでも起動できない場合は、サポート(help@affinity-science.com)までご連絡ください。
(2024/4/9)
Q10. Windows 7上でもYASARAを利用できますか?
Windows 7上でも動作しますが、もし、使用中に何か不具合が発生した場合は、弊社サポート(help@affinity-science.com)までご連絡ください。
(2024/5/9)
Q11. 金属イオンの結合部位を予測することができますか?
ShowIonSitesコマンドを使用することで可能です。現在対応しているのは、主に酸素原子に結合する陽イオンである、K+、Mg++、Ca++です。水分子が存在すると結果に影響することがあるため、事前に水分子を削除することが望ましいです。(メニューからEdit > Delete > Waters を選択するなど)
ShowIonSitesコマンドを実行するには、メニューから View > Show ion binding sites > All又はObjectを選択し、次に表示されるダイアログから金属イオンを選択します。結合部位が見つかった場合は、予測部位にドットが表示されます。小さくて見づらい場合があるので、メニューから Effects > Point&line appearance > Radius を選択し、ドットサイズを調整してください。コマンドコンソールからShowIonSitesコマンドを実行する方法もあります。詳細は、YASARAユーザーマニュアルの「ShowIonSites」コマンドページをご覧ください。
(2024/5/9)
Q12. 複数化合物のドッキングスクリーニングを実行するには?
ある一つの標的タンパク質に対し、複数のリガンドをドッキングさせるには、YASARA付属のマクロ「mcr」を使用します。実行手順については、YASARAユーザーマニュアルの
Recipes-Perform complex tasks > Dock a ligand to a receptor > Virtual screening of ligands the easy way
をご覧ください。
(2024/5/9)
Q13. アンサンブルドッキングとスクリーニングを同時にできるマクロファイルはありますか?
シミュレーションに時間が掛かりすぎてしまう可能性があるため、現在、アンサンブルドッキングと複数のリガンドのドッキングを同時にできるようなマクロファイルは提供されていません。代替手段としては、まずスクリーニング用マクロ「dock_runscreening.mcr」を実行し、続いて上位にヒットした構造に対して別途アンサンブルドッキング「dock_runensemble.mcr」を実行する方法があります。
(2024/5/9)
Q14. Bindenergy値は正に大きい方が結合が強いのですか?
YASARAにおけるBindEnergyは、「全体を別々のパーツに分解するのに必要なエネルギー」として扱われているため、正に大きいほど結合が強くなります。通常、YASARAにビルトインされたものではない、オリジナルのAutoDockなどでは、負に大きい方が結合が強いことを表しますが、YASARAでは符号が反転しますので、ご注意ください。なお、YASARAでAutoDockを指定してドッキングを実行した場合、単に符号が逆転しただけの結果が出力されます。論文などで報告する場合は、混乱を避けるためにYASARAで出力された符号を反転して記載することが推奨されています(例、10.916 を -10.916 として記載)。
詳細は、YASARAユーザーマニュアルの
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > BindEnergyObj-Calculate binding energies
をご覧ください。
(2024/5/9)
Q15. タンパク質のアミノ酸を置換や修飾することはできますか?
タンパク質の置換は「SwapRes」コマンドを使って実行することができます。このコマンドで置換できるアミノ酸としては、一般的な20種類のD-またはL-アミノ酸と、下記のプロトン化状態の異なる残基がサポートされています。
実行例を以下に記載します。
- メニューから実行する方法
-
- メニューからEdit > Swap > Residueを選択します。
- 表示されるダイアログから、置換前の残基を選択し、OKをクリックします。
- 続いて、次の画面から置換したい残基を選択し、OKをクリックします。
- 3Dビューから実行する方法
-
- 3Dビューから置換前の残基に含まれるいずれかの原子をクリックしてマークします。
- マークした原子を右クリックし、表示されるメニューからSwap > Residueを選択します。
- 表示されるダイアログから置換したい残基を選択し、OKをクリックします。
- Sequence Selectorから実行する方法
-
- 操作画面の下方にカーソルを動かし、Sequence Selectorを表示します。
- 置換前の残基名の上で右クリックし、表示されるメニューからSwap > Residueを選択します。
- 表示されるダイアログから置換したい残基を選択し、OKをクリックします。
- コマンドコンソールから実行する方法
-
- Spaceキーを押してコマンドコンソールを開きます。
- 「SwapRes (置換前の残基名),(置換後の残基名)」を入力し、Enterキーを押して実行します。
詳細については、YASARAユーザーマニュアルの「SwapRes」コマンドのページをご覧ください。
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > SwapRes-Swap residue side-chains
修飾されたアミノ酸を構築する方法については、YASARAユーザーマニュアルの「BuildRes」コマンドページ
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > BuildRes-Build single residue
内の、「Building unusual amino acids and nucleotides:~」以下をご参照ください。
(2024/5/9)
Q16. YASARAの新バージョンや別OSのインストーラを入手するには?
保守期間内であれば、次のサイトからいつでも新しいインストーラを入手することができます。
YASARA Update
https://www.yasara.org/update.htm
こちらの申請を行うには、Your email address or serial number:に「登録済の電子メールアドレス」または「ライセンスシリアル番号」、Update from YASARA version: にフルインストーラの場合「1.1.1」、差分アップデートの場合「Your previous download」、その他の項目は、リストからご希望のものをお選びいただき(最近のCPUであれば「64-bit, ~」 と「Include support for all processors, ~」で問題ないはずです)、最後に「Update…」ボタンを押します。
更新情報の入力例(フルパッケージ、WindowsとLinux、64-bit、Include support for all processorsを指定)
通常、数時間内に開発元より、新しいインストーラのダウンロード用URLが記された電子メールが登録済みの電子メールアドレス宛に送信されます。
なお、登録メールアドレスが2つあるグループリーダーライセンスをお持ちで、もう1人の方に送信されないようにする場合、シリアル番号の代わりにメールアドレスを入力してください。もしくは、次のページから受信者を設定することも可能です。
Administrate your Group Leader License
https://www.yasara.org/groupleader.htm
(2024/9/9)
Q17. オブジェクトやSceneの前後(画面の手前や奥)を不可視化して、中心部分をスライスして表示するような機能はありますか?
マウス操作にはそのような機能は割り当てられていませんが、「Cut」コマンドを使用することで似たような操作を行うことができます。
メニューから、View > Cut > All/Objectを選択し、表示オプションを選択(✓を入れたものがカットの対象となります)してOKをクリックするとカット面(Cut-Plane)オブジェクトが表示されます。
Cut-Planeが表示されたら、Shiftキーを押すか、画面右側SCENE CONTENTのCutPlaneをCtrl+左クリックして、Cut-Planeオブジェクトをマウスの操作対象にします。
マウスの右クリックを押しながらカーソルを動かすと、Cut-Planeの位置を調整できます。
なお、古いOpenGLなどでは表示オプションの設定が反映されない場合があるようです。そのような場合は、メニューのWindow > Renderer からレンダラーを変更してお試しください。
詳細については、YASARAユーザーマニュアルの「Cut」コマンドのページをご覧ください。
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > Cut<Obj|All>-Cut objects open
また、ヘルプムービーにはCut-Planeの項目があるので、こちらもご参照ください。
Help > Play help movie > 2.6. Cut-planes
(2024/9/19)
Q18. MDシミュレーションの実行時に、電界を設定することはできますか?
はい。AddESFコマンドを使用することで、現在の力場に静電場を追加することができます。
ユーザーマニュアルのコマンドページには、md_runmembrane.mcr中での設定例なども記載されていますので、詳細についてはそちらをご参照ください。
ユーザーマニュアルの「AddESF」コマンドページ
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > AddESF – Add electrostatic field
(2024/10/03)
Q19. 「Clean」や「OptHyd」コマンドを実行した際、意図しない箇所が自動的にプロトン化されてしまいます。
そのような場合は、以下のような対処法があります。ただし、その後マクロファイルを実行する場合は、マクロ内に「Clean」や「OptHyd」コマンドが含まれていないか確認し、pHの設定値を編集したり、自動で水素付加を行うコマンド(「Clean」「OptHyd」等)をコメントアウトするなどして適宜対応してください。
- pHを変更
水素付加は、現在設定されているpHに従って行われます。「pH」コマンド(メニューからはOptions > Default pH)で値を変更すると、Clean処理時のプロトン化状態が変化する場合があります。 - 不要な水素を削除
「Del」コマンドで不要なプロトンを削除するもできます。プロトンを削除すると、電荷も自動的に更新されます。結合次数も修正したい場合は、「AddBond」コマンドで変更できます。 - アミノ酸のプロトン化状態を変更したい場合は「SwapRes」コマンドを使用
「SwapRes」コマンドを使用することで、アミノ酸(アスパラギン酸、システイン、グルタミン酸、ヒスチジン)の側鎖のプロトン化状態を変更できます。 - Clean時に水素付加をスキップ
Cleanコマンドには、いくつかスキップパラメータがあり、オプションで「Skip=AddHyd」を指定することで水素付加の処理自体をスキップすることもできます。
(2024/10/03)
Q20. 自作の力場パラメータを使って、力場の修正ができますか?
はい。複雑なステップが必要ですが、力場パラメータを手動で定義することができます。
詳しくは、ユーザーマニュアルのRecipes > Run a molecular dynamics simulation > Preparing the force field の項目をご参照ください。
(2024/10/03)
Q21. プロテインモデリングの実行時に、機密配列が外部のサーバーに送信されますか。
いいえ。現行版(ver. 24.4.10)のYASARAでは、機密(ターゲット)配列は外部サーバーには送信されません。詳しくは、以下をご確認ください。
・Changelog(https://www.yasara.org/changelog.htm)より一部抜粋(ver. 24.4.10)。
(参考訳)新たなハイライト: YASARAに人工知能が登場。homology modeling experimentがprotein modeling experimentに名称変更され、AIベースのフォールディング手法AlphaFold、ESMFold、OmegaFoldと古典的なテンプレートベースのホモロジーモデリングを組み合わせてタンパク質構造を予測します。YASARAの基準に従って、お客様の潜在的な機密配列はローカルに保存され、外部サーバーには送信されません。詳細はwww.yasara.org/proteinfoldingをご覧ください。
・ユーザーマニュアルRecipes > Build a protein model > Protein modeling the easy wayより一部抜粋。
(参考訳)75GBの空きディスクスペースがあれば、pm_buildマクロのfoldmethod=’All’という行のコメントを外して、AlphaFoldを有効にすることができます。YASARAは、EBIに寄託されているすべてのAlphaFold予測の75GBの配列データベースをダウンロードし、ターゲット配列をそれとBLASTし、最も類似したAlphaFold予測(立体構造)をダウンロードし、テンプレートとして含めます。これは、あなたの潜在的な機密配列が外部に送信されないことを意味します。
(2024/10/03)
Q22. YASARAからAlphaFoldなどのAIベースのフォールディング手法を利用できますか。
YASARAでは、AlphaFold、Boltz-2、ESMFold、OmegaFold、といったAIフォールディング手法を利用することができます。
AlphaFoldに関しては、YASARA内で構造予測を行うのではなく、EBI(欧州バイオインフォマティクス研究所)のAlphaFold 配列データベースから、最適なモデルをBLAST 検索・ダウンロードして利用します。プロテインモデリング実行時にオプションを指定することで、テンプレート構造として利用することができます。
(ターゲット配列情報は外部に送信されませんが、構造データのダウンロードにインターネット環境が必要です)。
Boltz-2、ESMFold、OmegaFoldについては、構造予測プログラム本体(Python パッケージ)が YASARA に組み込まれて、YASARA を実行中の計算機単独で立体構造の予測が可能です(初回実行時に関連パッケージを自動ダウンロード、オフライン解析可)。
これらのAIフォールディング手法により容易された立体構造は、上述のAlphaFoldと同様にタンパクモデリング実行時のテンプレート構造として利用することができるほか、「FoldMol」「FoldObj」コマンドを実行して構造予測機能を単独で利用することもできます。
- 「FoldMol」コマンド:配列からタンパク質の構造予測を行うコマンド。対象は単量体のみで、Boltz-2、ESMFold、OmegaFoldによる立体構造予測が可能。
- 「FoldObj」コマンド:配列からタンパク質やDNA/RNA、リガンドを含む複合体構造全体の構造予測を行うコマンド。Boltz-2を利用。
詳しくはYASARAユーザーマニュアルの該当コマンドページをご参照下さい。
プロテインモデリングでのAIフォールディング手法の設定方法については、こちらで公開しているチュートリアル「タンパクモデリング」をご覧ください。
(2026/1/22)
Q23. macOSでのYASARAのインストール方法やアプリケーションの起動方法を教えてください。
- メールにてご案内したURLをクリックし、インストーラのダウンロードを行ってください。
- Finderで「InstallYASARA.zip」をダブルクリックして解凍します。もし自動で解凍されない場合は、ターミナルから「unzip」を使用してください。
- Finderを使用してYASARAアプリケーション(YASARA.app)を任意の場所(通常は/Applications)に移動します。
- ターミナルアプリを開き(Finderで「アプリケーション > ユーティリティ > ターミナル」をクリック)、以下のコマンドをコピーしてペーストし(/Applicationsにインストールしなかった場合は、パスを適宜変更してください)、’Return’を押してコマンドプロンプトが戻るまで数秒待ちます:
xattr -rc /Applications/YASARA.app && codesign --force --deep --sign - /Applications/YASARA.app - 上記のコマンドが実行できた場合は、YASARAを他のアプリのように開始できます。
エラーが発生した場合は、Cmd+AおよびCmd+Cを押してターミナルの出力をすべてコピーし、出力内容をお知らせください。
※こちらの内容は、YASARA version 24.10.5時の情報に基づいております。お問い合わせの際は、YASARAおよびmacOSのバージョン情報もあわせてご連絡ください。
(作成:2024/10/22、修正:2024/10/31)
Q24. マクロをYASARA GUIのコンソールに直接ペーストして実行することはできますか。
可能ですが、以下の2点に注意してください。
- 「if」「else」構文を含めない
マクロの各行が、コンソールに貼り付けられたときに即座に実行されてしまうため、これらの構文を含めることができません。 - ループが含まれる場合はループが完了している必要がある
ループが含まれる場合は、ループの後に必ず1つ以上のコマンドが存在する必要があります。コマンドが無い場合は、何も行わない「pass」コマンドを使用することができます。
例)for i=1 to Objects name = NameObj (i) SavePDB (i),(name) pass
参考:ユーザーマニュアル Macros-Automate your work with Yanaconda > Macros can be pasted directly into the console
(2025/8/1)
Q25. MDトラジェクトリファイル~.simを開くには?
MDシミュレーションを実行すると、作業ディレクトリにトラジェクトリファイル(~.sim)が作成されます。
~.simファイルを開くには、事前にMDシミュレーション実行時に作成される、溶媒が充填された初期構造ファイル「~_water.sce」を開いてから、メニューでFile > Load > Simulation snapshotを選択し、開きたいトラジェクトリファイルを指定して読み込みます。
参考:ユーザーマニュアル「LoadSim」コマンドページ
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > LoadSim – Load simulation snapshot in sim format
なお、YASARA付属のmd_play.mcrというマクロファイルを実行(マクロターゲットの指定が必要)すると、プレイヤーが起動し、全トラジェクトリを連続的に表示することもできます。プレイヤーを操作すると、構造の重ね合わせや、MP4形式で動画を出力することもできます。
参考:ユーザーマニュアル「Playing back a trajectory」ページ
Recipes-Perform complex tasks > Run a molecular dynamics simulation > Playing back a trajectory
(2025/8/1)
Q26. 相互作用の色の割り当ては?
YASARAで表示可能な主な相互作用とデフォルトの配色は以下になります。水素結合以外の相互作用は、相互作用の強さに合わせて色が変化するようになっているので、2色設定されています。デフォルトでは、強度が弱まるほどグレーに近づきます。
| 相互作用の種類 | 最小強度 | 最大強度 |
|---|---|---|
| 水素結合 | 黄色の点線 | 黄色の点線 |
| 疎水性 (Hydrophobic) | グレー | 緑 |
| π-π (Pi-pi) | グレー | 赤 |
| カチオン-π (Cation-pi) | グレー | 青 |
| イオン (Ionic) | グレー | マゼンタ |
参考:ユーザーマニュアル「ShowInt」コマンドページ
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > ShowInt<Atom,Res,Mol,Obj>-Show interactions
なお、水素結合以外の相互作用の配色は、メニューの View > Color > Parameters から変更することもできます。
参考:ユーザーマニュアル「ColorPar」コマンドページ
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > ColorPar-Set/get default color parameters
(2025/8/1)
Q27. 結合次数を確認する方法は?
結合の配色から確認する方法
- F2キーを押す(又はView > Style scene > Overall style: Ball&Stickを選択)
結合の色を反映できるStyleであるBall & Stickに変更します。 - View > Color > Bonds を選択し、表示されるダイアログ画面のName欄から「Order」を選択し、「OK」をクリック
すると、結合の棒が結合次数に応じた配色に変わります。 配色は以下になります。
コンソール画面に結合情報を出力して確認する方法
- Analyze > List > Bond を選択し、表示されるダイアログ画面で調べたい結合の両端の原子の1つ目を選択しOK、次の画面で2つ目の原子を選択しOK。
全ての結合の情報を出力したい場合は、右側の「Belongs to or has」リストから「All」を選択してください。 - 次のダイアログ画面では、リストアップする結合長の最小値を設定します。通常はデフォルト値(0 A)を使用します。(このオプションは欠損残基間をつなぐ長いペプチド結合をリストアップする場合などに使用します。)
- 画面下部のコンソール画面に結合情報のリストが出力され、結合次数が確認できます。
出力例
この操作で実行されるコマンドは、「ListBond」コマンドです。詳細はユーザーマニュアルのコマンドページをご覧下さい。
なお、結合次数はpHの影響を考慮して割り当てられます。FAQの「Q1. YASARAにおいて重要な10の単語(The ten magic words )」中に記載している【8】Bond の説明もご参照ください。
参考:
ユーザーマニュアル「ColorBond」コマンドページ
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > ColorBonds-Color bonds
ユーザーマニュアル「AddBond」コマンドページ
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > AddBond-Add covalent bonds
ユーザーマニュアル「ListBond」コマンドページ
Commands-Tell YASARA what to do > Index-All commands in alphabetic order > ListBond-List bonds
(2025/8/1)
Q28. MD解析レポートのグラフやプロットの数値データはどこから取得できますか?
MDトラジェクトの解析用マクロファイル「md_analyze」を実行すると、様々なデータファイルが作成され、作業フォルダに保存されます。
このうち、「(MacroTarget)_analysis.tab」というファイルに、解析レポート内の各プロットや表の数値データの結果が保存されています。
「md_analyze」マクロ実行時に作成される解析ファイルの種類や内容については、解析レポート「(MacroTarget)_report.html」の末尾に記載されているほか、レポート中の各プロットや表の説明の中に関連ファイル名が記載されている場合もあります。
また、YASARAユーザーマニュアルの以下の項目に、作成されるファイルの説明などが記載されていますので、併せてご参照ください。
ユーザーマニュアル該当箇所:Recipes > Run a molecular dynamics simulation > Analyzing a trajectory
(2026/1/22)